SimulateReactivity
SimulateReactivity[initialState]⟹reactionMechanism
generates a putative ReactionMechanism that describes the hybridization behavior of the system of nucleic acids or adds reaction rates to known reaction types from in the nucleic acid.
SimulateReactivity[reactionMechanism]⟹reactionMechanism
adds reaction rates to known reaction types from in the nucleic acid reactionMechanism.
Details
- SimulateReactivity looks for possible reactions of following types: Folding/Melting, Zipping/Unzipping, Hybridization/Dissociation, StrandInvasion, DuplexInvasion, ToeholdMediatedStrandExchange, ToeholdMediatedDuplexExchange and DualToeholdMediatedDuplexExchange. Exhaustive searching is used for finding reactions, i.e. each species is simulated for first order reactions ane each pair of species is simulated for second order reaction. SimulateKinetics is used to exclude unfavorable structures.
- When given a state containing strands with sequences, SimulateReactivity uses Base method by default, which considers Watson-Crick rules to look for valid reactions.
- When given a state containing strands without sequences, SimulateReactivity uses Motif method, which considers motif matching to find possible products and use kinetic simulation to determine strong reactions.
- Theoretical polymerization can be avoided by setting the Depth option to a small number. Theoretical polymerization can also be excluded by turning on PrunPercentage option.
- When a temperature is given, SimulateReactivity will calculate reaction rates with KineticRates function.
- Rate for Folding is considered dependent on temperature and monovalent salt concentration as described by Bonnet, G., Krichevsky, O., & Libchaber, A. (1998) in Object[Report,Literature,"id:E8zoYvNkERq5"]: "Kinetics of conformational fluctuations in DNA hairpin-loops." Proceedings of the National Academy of Sciences, 95(15), 8602-8606. The folding rate is fitted by using the kinetics data as a function of 1000/T (K^-1) and monovalent salt concentration (Molar) as described in Object[Analysis,Fit,"id:AEqRl9Kz9YKw"].
- Rate for Hybridization is dependent on monovalent salt concentration as decribed by James G. Wetmur (1991) in Object[Report,Literature,"id:wqW9BP7kDBKV"]: "DNA probes: applications of the principles of nucleic acid hybridization." Critical reviews in biochemistry and molecular biology, 26(3-4), 227-259. In the range of interest ([Na+] >= 0.25 Molar), the hybridization rate is given by Kon = {4.35*Log10[Na+]+3.5}*10^5. When salt concentration is below 0.25 Molar, hybridization starts becoming more stringent as only perfectly matched hybrids will be stable, thus the rate drops according to decreasing salt concentration. Temperature also plays a vital role in hybridization rate as described by Chunlai Chen, et al (2007) in Object[Report,Literature,"id:BYDOjvGNmwNr"]: "Influence of secondary structure on kinetics and reaction mechanism of DNA hybridization" Nucleic acids research, 35(9), 2875-2884. Log[Kon] was fitted as a function of 1/RT (mol*K/cal) where R is the ideal gas constant, the fitted function as described in Object[Analysis,Fit,"id:01G6nvw7veVE"] is used to predict the hybridization rate given the temperature.
- Rate for ToeholdMediatedStrandExchange uses kinetic data described in Object[Report, Literature, "id:XnlV5jmalqMZ"] : B. M. Frezza "Orchestration of molecular information through higher order chemical recognition." (2010). This data was log-normalized and fitted to compute kinetic rates as function of the activation energy normalized by the Temperature and Boltzmann factor in Object[Analysis, Fit, "id:dORYzZJKnMd5"] for 3' toehold and Object[Analysis, Fit, "id:P5ZnEjdL4nkE"] for 5' toehold.
- Rate for ToeholdMediatedDuplexExchange uses kinetic data described in Object[Report, Literature, "id:XnlV5jmalqMZ"] : B. M. Frezza "Orchestration of molecular information through higher order chemical recognition." (2010).
- Rate for DualToeholdMediatedDuplexExchange is similar to ToeholdMediatedStrandExchange but with a different AnalyzeFit object as in Object[Analysis,Fit,"id:mnk9jORoVr9w"].
- Rate for StrandInvasion uses kinetic data described in: L. P. Reynaldo et al in Object[Report,Literature,"id:7X104vnRe98J"]: "The kinetics of oligonucleotide replacements." J. Mol. Biol. 297 (2000): 511-520. For strand invasion mediated by the sequential displacement pathway, related kinetic data was fitted as a function of the number of displaced bonds and the temperature in Object[Analysis,Fit,"id:Vrbp1jKDZZzx"]. For strand invasion mediated by the dissociative pathway, related kinetic data was fitted as a function of the number of dissociated bonds and the temperature in Object[Analysis,Fit,"id:9RdZXv1W7E9L"].
- Rate for DuplexInvasion is as described in Object[Report, Literature, "id:XnlV5jmalqMZ"] : B. M. Frezza "Orchestration of molecular information through higher order chemical recognition." (2010), signal measured was not significant. Thus the duplex invasion rate is set to be 0/Molar/Second.
Input
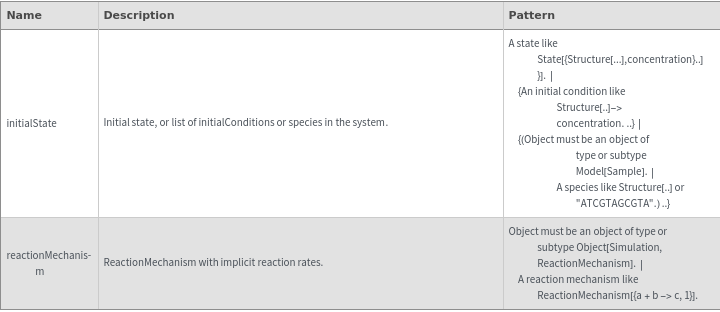
Output
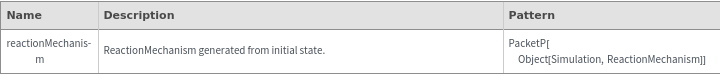
General Options
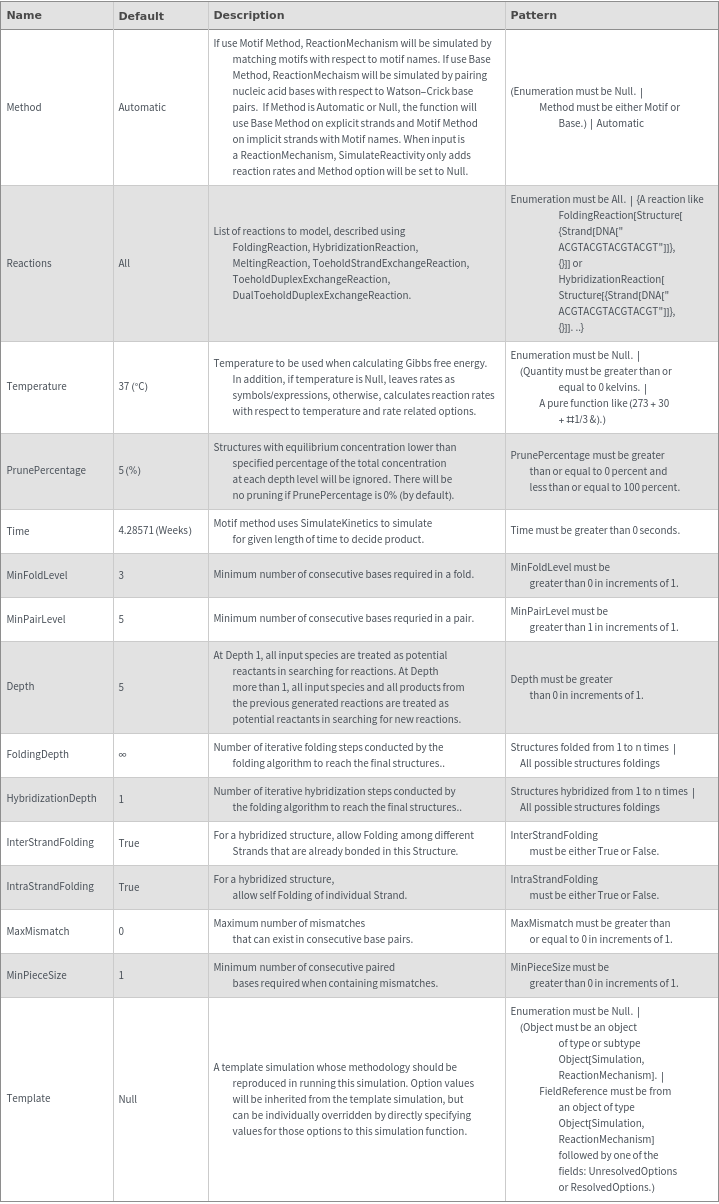
Examples
Basic Examples (5)
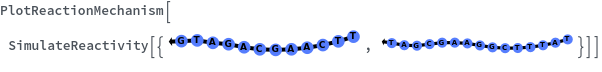

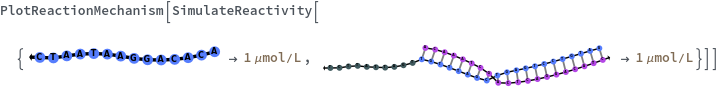


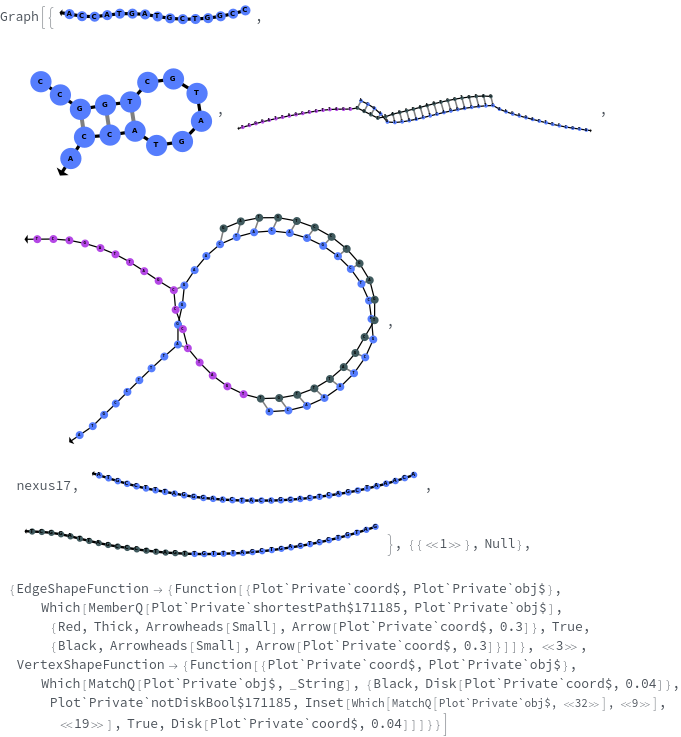



Options (19)
Depth (2)
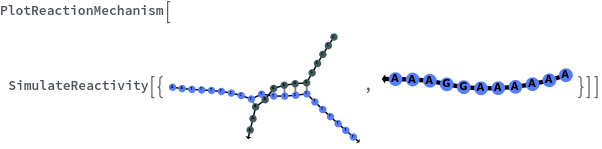
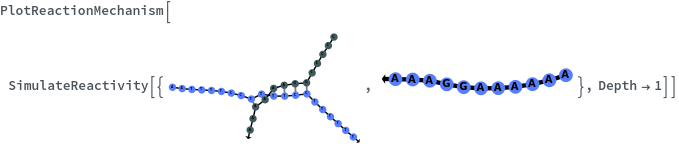
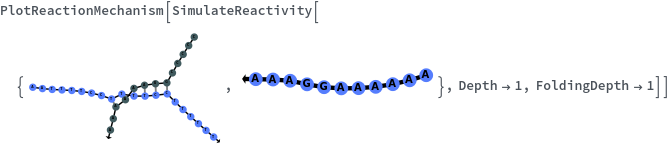
HybridizationDepth (1)

InterStrandFolding (1)
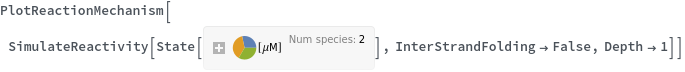
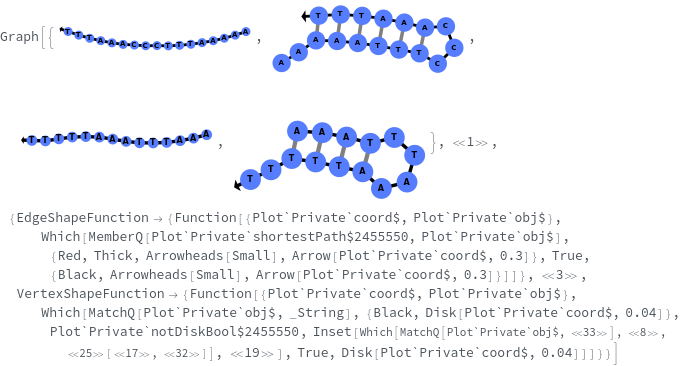
IntraStrandFolding (1)
MaxMismatch (2)



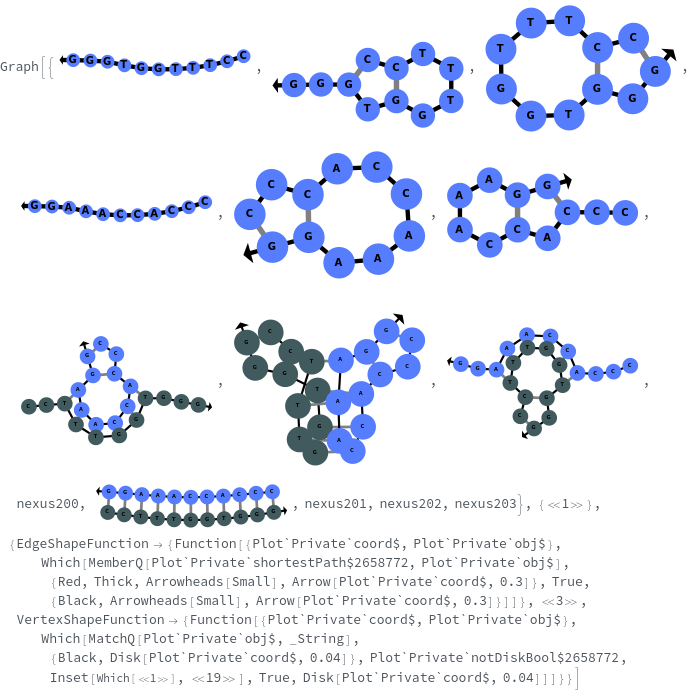
Method (2)

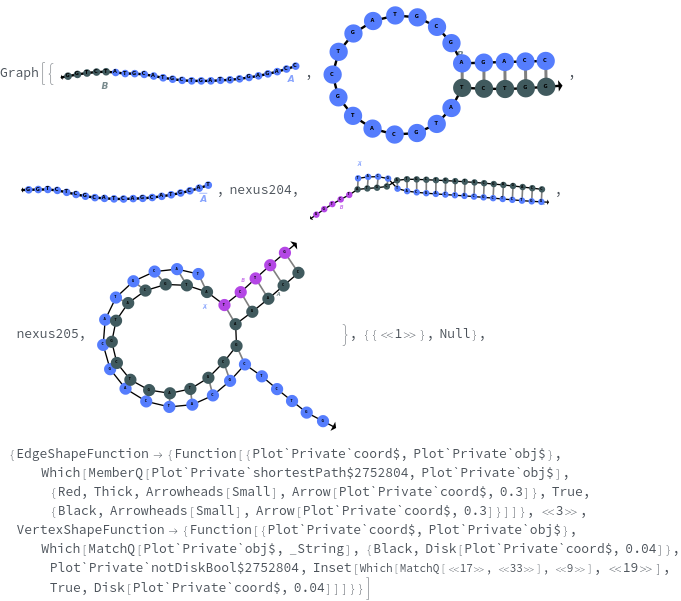





MinPieceSize (1)

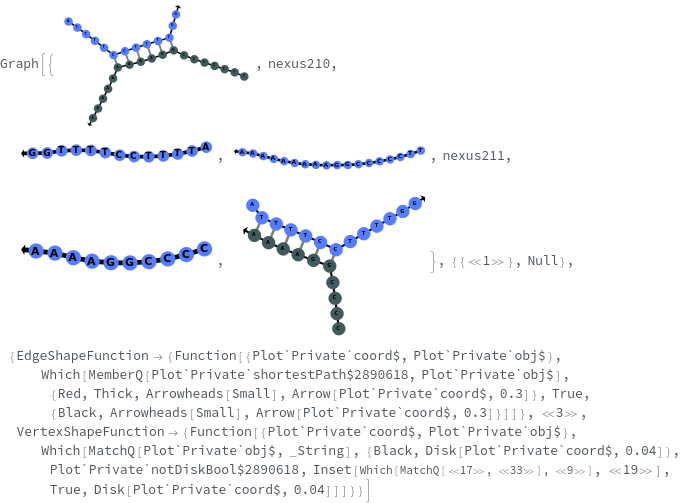

Template (2)
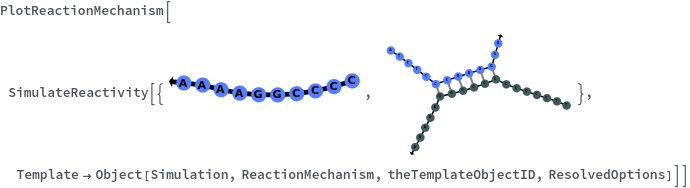

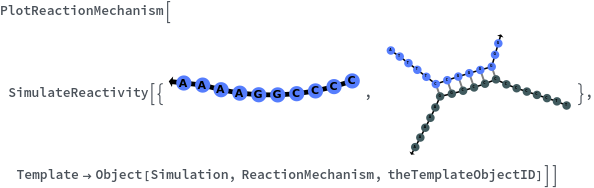



Messages (2)
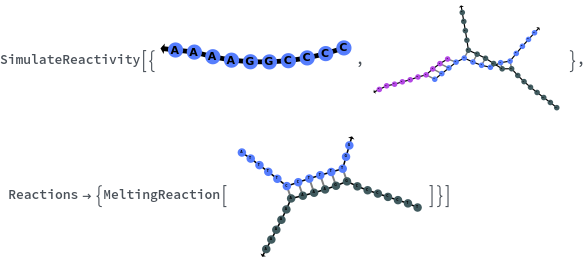


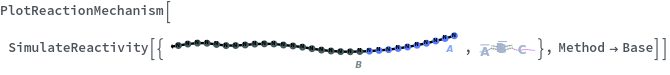


Applications (2)
Generate a ReactionMechanism from initial state then call SimulateEquilibrium to simulate the equilibrium state of the ReactionMechanism:

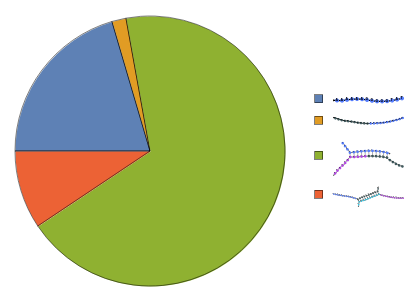
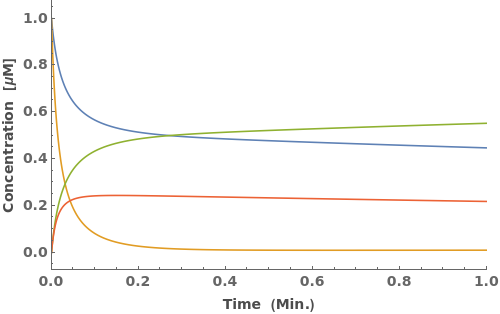
Instead of simulate equilibrium state directly, you can also call SimulateKinetics to simulate the kinetic trajectory of the ReactionMechanism to see concentration changes with respect to time: